Alfa-mannosidose er en sjelden, recessivt arvelig, lysosomal sykdom forårsaket av mutasjoner i MAN2B1 (LAMAN) genet på kromosom 19. Personer med sykdommen mangler helt eller delvis funksjonen til enzymet alfa-mannosidase, noe som fører til opphopning av mannoserike oligosakkarider i lysosomene, med forstyrrelse av cellefunksjon, samt apoptose som konsekvens. 1,2 Uttrykket av MAN2B1 er høyest i lunger, nyrer, bukspyttkjertel og leukocytter. I CNS er uttrykket høyest i hjernestammen og ryggmargen.1
Alfa-mannosidose er en livslang, multisystemsykdom med ofte langsom, men jevn forverring av for eksempel skjelett- og nevromuskulære symptomer.1 Sykdommen har en høy grad av fenotypisk variasjon, som debutalder og alvorlighetsgrad, hvor forskjeller sees selv blant søsken med identiske genotyper.1, 3
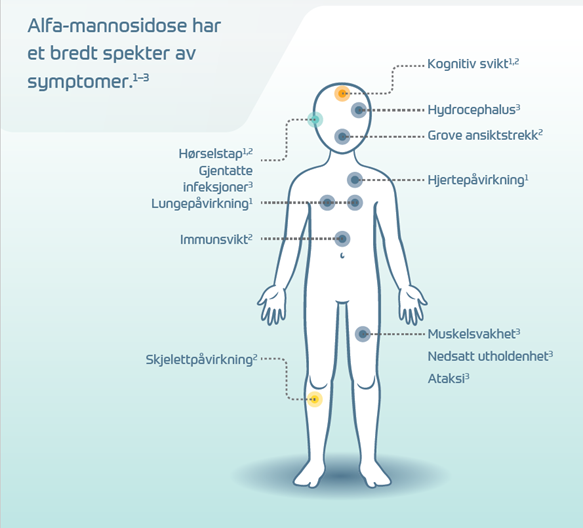
De fleste pasienter med alfa-mannosidose har nedsatt hørsel allerede i tidlig alder og tilbakevendende infeksjoner i ørene og øvre luftveier. De utvikler ofte et karakteristisk utseende som blir mer fremtredende med alderen.3 Andre vanlige symptomer er redusert muskeltonus, ataksi og intellektuell svikt.1,2 Over tid kan pasienten oppleve ytterligere nedsatt bevegelighet, smerter i skjelettet og psykiske problemer.1, 3
Riktig diagnose er av stor betydning for valg av behandling og for at pasienten tidlig skal få kontakt med nødvendige spesialister. Ved symptomatisk hørselstap, med eller uten utviklingshemming, bør prøve sendes til genetisk avdeling for helgenomscreening eller genetisk screeningpaneltesting som inneholder MAN2B1-genet.1,4
Det finnes for tiden ingen kur for alfa-mannosidose, og behandlingen er ofte begrenset til å håndtere eller kontrollere symptomene som oppstår, samt forebygge medisinske komplikasjoner og kompensere for funksjonssvikt.1
For pasienter med mild til moderat form av sykdommen er det mulighet for enzymerstatningsterapi (ERT) for å behandle de ikke-nevrologiske manifestasjonene.5-6 Tidlig hematopoetisk stamcelletransplantasjon har for noen pasienter vist seg å ha en positiv effekt ved å stanse utviklingen av sykdommen.7
Referanser:
- Malm, D., Nilssen, Ø. Alpha-mannosidosis. Orphanet J Rare Dis 3, 21 (2008). https://doi.org/10.1186/1750-1172-3-21
- Borgwardt L, Stensland HM, Olsen KJ, et al. Alpha-mannosidosis: correlation between phenotype, genotype and mutant MAN2B1 subcellular localisation. Orphanet J Rare Dis. 2015;10:70. Published 2015 Jun 6. doi:10.1186/s13023-015-0286-x
- Beck M, Olsen KJ, Wraith JE, et al. Natural history of alpha mannosidosis a longitudinal study. Orphanet J Rare Dis. 2013;8:88. Published 2013 Jun 20. doi:10.1186/1750-1172-8-88
- Guffon N, Tylki-Szymanska A, Borgwardt L, et al. Recognition of alpha-mannosidosis in paediatric and adult patients: Presentation of a diagnostic algorithm from an international working group. Mol Genet Metab. 2019;126(4):470-474. doi:10.1016/j.ymgme.2019.01.024
- Borgwardt L, Guffon N, Amraoui Y, et al. Efficacy and safety of Velmanase alfa in the treatment of patients with alpha-mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double-blind, randomised, placebo-controlled trial. J Inherit Metab Dis. 2018;41(6):1215-1223. doi:10.1007/s10545-018-0185-0
- Harmatz P, Cattaneo F, Ardigò D, et al. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): Novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol Genet Metab. 2018;124(2):152-160. doi:10.1016/j.ymgme.2018.04.003
- Ceccarini MR, Codini M, Conte C, et al. Alpha-Mannosidosis: Therapeutic Strategies. Int J Mol Sci. 2018;19(5):1500. Published 2018 May 17. doi:10.3390/ijms19051500