Fabrys sykdom er en sjelden, X-bundet, lysosomal lidelse med en forekomst på mellom 40 000–117 000.2 Sykdommen er forårsaket av mangel på enzymet α-galaktosidase A, som fører til akkumulering av globotriaosylceramid (Gb3) og andre relaterte glykosphingolipider, i de fleste celletyper i kroppen. Sykdommen påvirker mange organer og vev i kroppen som nyrer, hjerte og nervesystem.1
Alder ved symptomdebut varierer og kan opptre tidligere hos menn enn hos kvinner.3 Fabrys sykdom kan deles inn i klassisk sykdom, som oftest ses hos menn uten gjenværende enzymnivå, og ikke-klassisk sykdom, som generelt er mindre alvorlig og preget av et mer varierende sykdomsforløp.1 Diagnostisering av klassisk Fabrys sykdom hos menn kan være enkel, mens diagnose hos kvinner og hos individer med genetiske varianter oppleves som mer utfordrende.4 Symptomene på Fabrys sykdom kan variere mye og noen ganger være diffuse. På grunn av dette, og fordi denne sykdommen er sjelden, kan tegn på Fabrys sykdom forveksles med andre sykdommer og undersøkelsene kan noe ganger ta flere år.5 Diagnosen er forskjellig mellom kjønnene; hos menn med typiske symptomer og/eller kjent arvelighet er det tilstrekkelig å måle mengden alfa-galaktosidase A, mens det for kvinner, som kan ha normale nivåer av enzymet, utføres en DNA-analyse av GLA-genet.6, 7
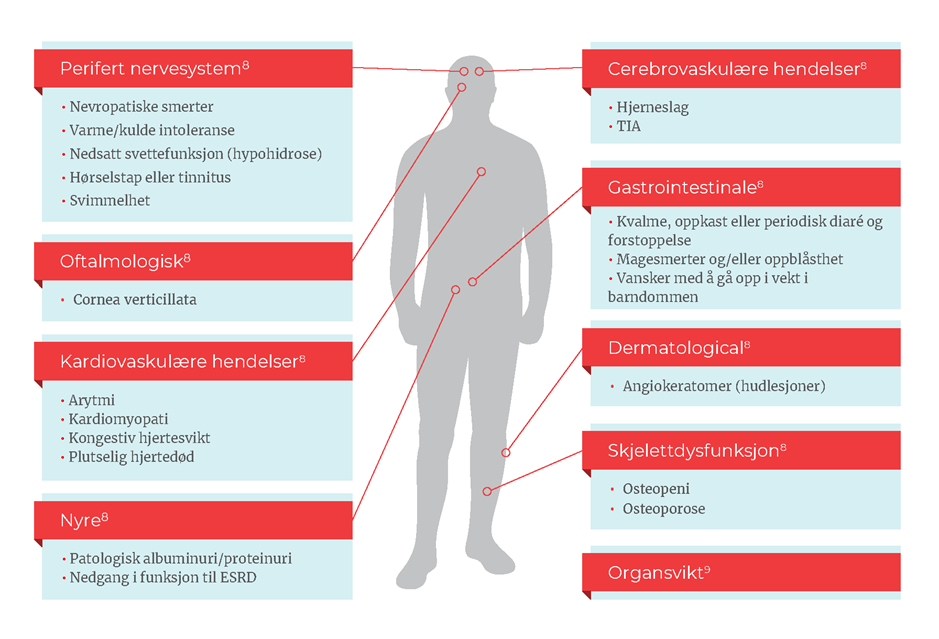
Viktigheten av tidlig diagnose
Behovet for rask og nøyaktig diagnose av denne progressive sykdommen er kritisk slik at pasienter kan identifiseres og behandles før de får irreversibel organskade.10 Ideelt sett bør behandlingen lindre de tidlige symptomene og redusere eller eliminere risikoen for å utvikle livstruende komplikasjoner senere i livet.3

Referanser:
- Arends M, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. 2017;28(5):1631–1641;
- Waldek S, et al. Life expectancy and cause of death in males and females with Fabry disease: Findings from the Fabry Registry. Genet Med. 2009;11(11):790–796;
- Hopkin RJ, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res. 2008;64:550–555;
- Vardarli I, et al. Diagnosis and screening of patients with Fabry disease. Ther Clin Risk Manag. 2020;16:551–558;
- Oslo Universitetssykehus: https://www.oslo-universitetssykehus.no/behandlinger/fabry-sykdom/#tegn-og-symptomer (Last accessed 26.02.2025)
- WebMD LLC: https://www.webmd.com/a-to-z-guides/fabry-disease-20/fabry-disease-genetics (Last accessed 26.02.2025)
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. Published 2010 Nov 22. doi:10.1186/1750-1172-5-30
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–427;
- Weidemann F, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. 2013;274(4):331–341;
- Desnick RJ, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338–346;